Principally, the primary function of
erythropoietin is to induce erythropoiesis, red blood cell production, in
hematopoietic tissues but also to maintain hemoglobin concentrations within a
normal range during steady-state conditions (Hadley & Levine, 2007). The
release of erythropoietin is stimulated by a reduction of oxygen (O2)
content in the blood (Hadley & Levine, 2007). EPO is essential for the
proliferation and survival of erythrocyte progenitor (erythroid) cells in the
bone marrow or fetal liver (Ng et. al, 2003). Furthermore, the glycoprotein has
also been implicated in having immunomodulatory effects in the body (Ng et. al,
2003).

Figure 1: EPO action on erythroid cells of
hematopoietic bone marrow.
{kind=link}
Studies have shown that circulating EPO
increases exponentially following a sharp decrease in hemoglobin levels, which
may occur in the case of ischemia (Hadley & Levine, 2007). The feedback
regulator of EPO production however, is not mediated by hemoglobin
concentration or red blood cell concentration in the body but by oxygen
pressure in the tissues (pO2) (Hadley & Levine, 2007). Tissue
oxygen pressure therefore depends on hemoglobin concentration which corresponds
to erythrocyte level in the body (Silverthorn, 2009). It is because of this
feedback mechanism that habitation at higher altitudes, where atmospheric oxygen
pressure is low, EPO production is stimulated to increase the production of red
blood cells and hence the carrying capacity of oxygen to the body (Silverthorn,
2009).

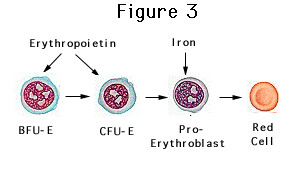
Figure 3: Pictographic description of the
primary function of erythropoietin in the body.
It is postulated that oxygen sensing in the
body is carried out by a heme protein, however recent evidence implicate that reactive
oxygen species (ROS) triggers the hypoxia-induced transcription of the EPO gene
(Hadley & Levine, 2007). The hypoxia-induced factor (HIF) is a family of proteins
which serve as transcriptional factors to mediate oxygen homeostasis (Hadley
& Levine, 2007). HIF-2 is the thought to be the major transcriptional
factor that activates EPO gene expression (Hadley & Levine, 2007).
When erythrocyte level is low in the body,
peritubular fibroblasts in the kidneys are stimulated and release
erythropoietin into the peritubular capillaries which transports the hormone
into the general systemic circulation. EPO then enters the vascular system
which supports the hematopoietic bone marrow (red marrow) and binds to specific
erythroid precursor cells that expresses the EPO receptor (Ng et. al, 2003).
The erythropoietin receptor is a member of the superfamily of cytokine
receptors (e.g. GH receptor) and characterized as a 72-78 kDa glycosylated/
phosphorylated transmembrane polypeptide (Ng et. al, 2003). There are two
distinct erythroid progenitor cells contained within the erythroid cell
compartment of the bone marrow; the burst forming unit-erythroid (BFU-E), and
the colony forming unit-erythroid (CFU-E) (Fisher, 2010). Initially, the BFU-E
does not appear to be sensitive to EPO but gains sensitivity as it matures,
whereas the CFU-E is inherently a more mature cell and serves as the primary
target of EPO (Fisher, 2010). CFU-E differentiation is completely dependent on
erythropoietin.
Binding of EPO to its receptor in the membrane of erythroid cells
induces a dimerization of the receptor with another erythropoietin receptor
(homodimer), causing a conformational change that allows Janus family tyrosine
protein kinase 2 (JAK2) molecules to associate with the receptor and become
activated by trans-phosphorylation (Fisher, 2010). JAK2 molecules in turn
phosphorylate tyrosine residues of the EPO receptor in the cytoplasmic domain
(Fisher, 2010). These residues serve as a docking site for various homologous
intracellular proteins which leads to the activation of STAT5A and STAT5B, as
well as the Ras/MAP kinase pathway and other kinases, all of which are involved
in gene activation (Fisher, 2010). The STAT5A and STAT5B molecules associate
following activation and translocate to the nucleus where it binds specific
hormone response elements that leads to transcription and translation of
proteins which triggers cellular proliferation (Fisher, 2010). The acute of effects
of EPO lasts between 30 and 60 minutes (Hadley & Levine, 2007).

Figure 4: Simplistic diagram of the main
signal transduction pathways activated by EPO receptor.
Termination of EPO’s effects is accomplished
by a hematopoietic cell phosphatase (HCP) which catalyzes the
de-phosphorylation of JAK2 (Hadley & Levine, 2007). Mutations of the HCP
enzyme or the erythropoietin receptor leads to erythrocytosis, an abnormally
high red blood cell level (Hadley & Levine, 2007).
Erythropoietin exerts several
non-erythropoietic actions including stimulation of mitosis along with
cardioprotective and neuroprotective effects (Fisher, 2010). EPO increases the
number of endothelial progenitor cells, as well as migration of the mature
forms (Hadley & Levine, 2007). Hepcidin suppression by EPO also increases
iron absorption in the body, which is a required building block for
erythrocytes (Silverthorn, 2009). A complete knock out of the EPO receptor gene
results in severe cardiac malformations and fetal death, possibly due to tissue
hypoxia and anemia (Hadley & Levine, 2007).
It has been demonstrated that EPO can act as a neuroprotective whereby it inhibits the death of neurons due to lack of blood supply and oxygen (Fisher 2010). This is accomplished by preventing the formation of oxygen radicals due to nitrogen oxide accumulation, or by antagonizing the effects of these radicals (Fisher, 2010). Preliminary studies have also shown that recombinant human EPO produces a protective effect in animal models for multiple sclerosis (Fisher 2010). In addition, EPO may act to inhibit apoptosis in cardiomyocytes and reduce the deterioration of these cells following an ischemic event, such as a myocardial infarction (Fisher, 2010). It has been postulated that non-hematopoietic effects of EPO may be regulated by heteromers of the erythropoietin receptor (Hadley & Levine, 2007).
It has been demonstrated that EPO can act as a neuroprotective whereby it inhibits the death of neurons due to lack of blood supply and oxygen (Fisher 2010). This is accomplished by preventing the formation of oxygen radicals due to nitrogen oxide accumulation, or by antagonizing the effects of these radicals (Fisher, 2010). Preliminary studies have also shown that recombinant human EPO produces a protective effect in animal models for multiple sclerosis (Fisher 2010). In addition, EPO may act to inhibit apoptosis in cardiomyocytes and reduce the deterioration of these cells following an ischemic event, such as a myocardial infarction (Fisher, 2010). It has been postulated that non-hematopoietic effects of EPO may be regulated by heteromers of the erythropoietin receptor (Hadley & Levine, 2007).

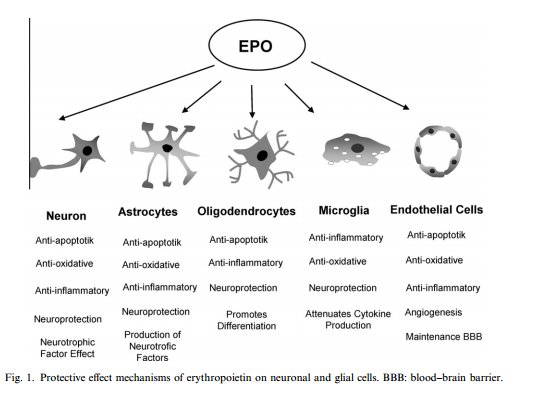
Figure
5: Non-hematopoietic effects of EPO on cells of the nervous system.
{kind=link}

Figure 6: (a) Coronal section of mouse brain
following blunt force trauma and treated with saline solution(above) compared
to coronal section of mouse brain following similar trauma but treated with
recombinant human erythropoietin (below). (b) Bar graph depicting the relative
degree of necrosis, following blunt force damage, in the brain region in a sham
control and a EPO treated subject.
Chronic renal failure can lead to anemia,
lower than normal red blood cell levels, which is caused by an insufficient
amount of EPO required for new erythrocyte production (Fisher, 2010). This
insufficiency is perpetuated by a decrease in renal function. Another proposed
cause is resistance to endogenous EPO (Fisher, 2010). Studies on mice have
shown that a knockout of the EPO gene during embryological development is lethal,
caused by anemia and heart hypoplasia, therefore the same knockout effect in
adults can only be studied using a gene silencer such as Cre recombinase
(Zeigler et. al, 2010). Following induction of Cre, mutant mice exhibited a
much lower serum EPO level and consequently developed chronic, normocytic,
anemia (Zeigler, 2010). It was also demonstrated that target genes, which is
acted upon by EPO, is significantly reduced in the bone marrow. These observations
are similar to the clinical signs of chronic kidney disease (Zeigler, 2010).
Systemic overexpression of EPO results in erythrocytosis and the subsequent high erythrocyte level increases the hematocrit. However, drugs are currently under development that will act enhance the protective effects of EPO without increasing the hematocrit (Jelkmann, 2007).
Systemic overexpression of EPO results in erythrocytosis and the subsequent high erythrocyte level increases the hematocrit. However, drugs are currently under development that will act enhance the protective effects of EPO without increasing the hematocrit (Jelkmann, 2007).

Figure 7: Hematocrit levels for different
levels of erythrocytes; affected by EPO conc.
References
- Hadley, M. E. & Levine, J. E. (2007).
Endocrinology (6th ed.). Prentice Hall, Pearson Education: Upper
Saddle River, NJ
-
Fisher, J. W. (2010) Landmark advances in the
development of erythropoietin. Experimental Biology and
Medicine. 235 (12): 1398-1411
-
Ng, T., Marx, G., Littlewood, T., & Macdougall, I. (2003) Recombinant
erythropoietin in clinical practice, Postgraduate
Medical Journal, 79: 367-376
-
Silverthorn, D. U. (2009). Human
Physiology, An integrated approach
(5th ed.). Benjamin Cummings, Pearson Education : San Francisco, CA
-
Jelkmann, W. (2007). Erythropoietin after
a century of research: younger than ever. European Journal of
Haematology. Institute of Physiology,
University of Luebeck: Luebeck, Germany
- Zeigler, B.M. Vajdos, J. Qin, W. Loverro,
L. Niss, K. (2010) A mouse model for an erythropoietin-deficiency anemia, Disease models and mechanisms, 3(11-12);
763-772
No comments:
Post a Comment