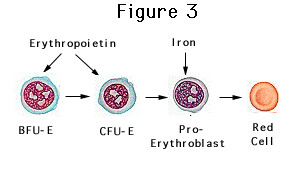
Although the primary role of erythropoietin
is to stimulate erythropoiesis, it has been shown to have a multitude of
non-erythropoietic effects (Fisher, 2010). One such effect includes a
cardioprotective mechanism that is activated by EPO during onset of cardiac
ischemia (Hirata et. al, 2006). Ischemia refers to the restriction of blood
flow to the tissues which in turn restricts the oxygen and glucose supply to
these tissues, needed for cell metabolism (Silverthorn, 2009). A supplementary
function of EPO during ischemia is the stimulation of endothelial progenitor
cell (EPC) mobilization which is predicted to enhance neovascularization of
ischemic regions (Hadley & Levine, 2007). “Erythropoietin Enhances
Neovascularization of Ischemic Myocardium and Improves Left Ventricular
Dysfunction after Myocardial Infarction in Dogs” is a journal article that
pertains to a study conducted by Hirata et. al (2006), which aims to
characterize the actions of erythropoietin on neovascularization and cardiac
function following a myocardial infarction (heart attack). The investigators
hypothesized that erythropoietin increases blood supply through ischemic
regions by neovascularization and improving cardiac function after an ischemic
event (Hirata et. al, 2006). The subsequent experiments were performed on
beagle dogs due to similar cardiovascular homology to humans.
(A)

(B)
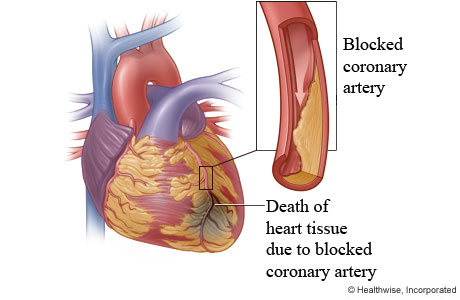
Figure 1
(A) & (B): (A) Adult grown Beagle dog. (B) Simplistic diagram depicting
ischemia induced myocardial infarction (heart attack).
Images
Retrieved from:
In this particular study, the investigators
tested the acute effects that EPO administration has on the size of a
myocardial infarction and also the effects of immediate or delayed EPO
administration on neovascularization of the cardiac tissue and the resulting
function.
In the immediate/delayed effects test, the investigators set up a control group (consisting of 8 dogs) and three experimental groups which received EPO at different time points following the LAD ligation. This enables the researchers to determine both short term and long term effects in one test. Similarly, myocardial blood flow and infarct size was measured at relevant time points. Cardiac function was determined using echocardiography, and tissue samples from both ischemic and non-ischemic regions were obtained and compared after all tests were conducted.
For both experiments, hemodynamic parameters
were measured at all relevant time intervals to monitor mean arterial blood
pressure, heart rate, and left-ventricular end diastolic pressure.
(A)

(B)

Figure 2:
(A) Measurement protocols to determine acute effects of EPO administration on
infarct size. (B) Measurement protocols to determine effects of immediate and
delayed administration of EPO on neovascularization and cardiac function.
Results Summary
Acute Effects Test;
- According to Figure 3, the researchers
demonstrated that the myocardial infarct size was observed to be significantly
smaller in dogs treated with EPO than the control group, treated with saline
(Hirata et. al, 2006). However, there was little difference observed between
the two groups regarding regional blood flow to the heart, and also the area
affected by infarct-induced necrosis (tissue death). Referring to figure 3A and
3B, necrosis (blackened tissue) was present to the same degree in both control
and EPO-treated groups (Hirata et. al, 2006).
.jpg)
Figure 3:
(A) Left ventricular cross section of heart, treated with saline, at 6 hours
following heart attack. (B) Left ventricular cross section of heart, treated
with EPO, at 6 hours following heart attack. (C) Graph depicting infarct size
at 6 hours, following heart attack, for control group and EPO-treated group
(open circles indicate infarct size of individual animals).
EPO administration immediately following LAD
ligation improved cardiac function within 90 minutes after the heart attack
event, presumably because of reduced infarct size which prevented long-term
dysfunction to develop afterwards (Hirata et. al, 2006).
Immediate/Delayed Effects Test;
The vascular endothelial growth factor (VEGF)
levels in the blood was shown to be elevated in both the control and EPO (0 hr)
groups, peaking around 6 hours following heart attack (Hirata et. al, 2006).
Therefore, this finding indicates that EPO administration did not affect VEGF
levels. It has been shown that both VEGF and EPO stimulate endothelial
progenitor cell (EPC) mobilization and that these two factors may act
synergistically (Hirata et. al, 2006). However, it is possible that the dose of
EPO used for testing was not sufficiently high enough to promote VEGF
proliferation and subsequent endothelial cell production, and it is further
postulated that higher doses of EPO would produce different results (Hirata et.
al, 2006).
According to Figure 4, there was no
significant difference in the LCX (non-ischemic) region regarding capillary
density and capillary to myocyte ratio when comparing the groups. However, in
the LAD (ischemic) region both the capillary density and capillary to myocyte
ratio was much higher in the EPO (0 hr) and EPO (6 hr) groups but not in the
EPO (1 wk) group (Hirata et. al, 2006). This finding suggests that, during
acute phase, EPO promotes neovascularization in the ischemic region. The
mechanism by which this occurs is thought to be accomplished by mobilizing
endothelial cells, and increases the amount of blood flow to the ischemic
region by increasing the number of capillaries feeding into the region (Hirata
et. al, 2006).

Figure 4:
(A) Images depicting non-ischemic regions (LCX region, a-d) and ischemic
regions (LAD region, e-h) in the control and experimental groups, using
immunohistologic staining with an antibody. (B) Graph depicting capillary
density in LCX and LAD regions. (C) Graph depicting capillary to myocyte ratio
in LCX and LAD regions.
Graph A from Figure 5 depicts the relative
number of mononuclear cells (CD-34 positive) detected within circulation. The
number of mononuclear cells (MNC’s) increased in all groups after 1 week of
heart attack (Hirata et. al, 2006). The EPO (0 hr) and EPO (6 hr) groups
exhibited significantly higher levels of MNCs compared to the control group and
EPO (1 wk) group. Similarly, at 2 weeks after heart attack event the MNC level
for control group and EPO (1 wk) returned to baseline levels, whereas MNC
levels for EPO (0 hr) and EPO (6 hr) groups remained high. All groups returned
to baseline levels after 4 weeks of the heart attack event (Hirata et. al,
2006). MNC count was shown to correlate with endothelial cell number and that
increased levels of MNC by EPO administration, indirectly relates to elevated
endothelial cells.

Figure 5:
(A) Changes of circulating mononuclear cells (CD-34 positive) after LAD
ligation in control group and experimental groups.
According to Figure 6, there was no pertinent
difference in myocardial blood flow to the ischemic region between any of the
groups, at 90 minutes post heart attack event (Hirata et. al, 2006). However,
at 4 weeks post heart attack event the blood flow increased markedly in the EPO
(0 hr) and EPO (6 hr) groups but not in the control group or the EPO (1 wk)
group (Hirata et. al, 2006). Because neovascularization was enhanced in both of
these groups as well, it is thought that the increased blood flow maybe a secondary
effect to the increased capillary density.

Figure 6: Trend
of myocardial blood flow to the ischemic region (LAD region) at 90 mins to 4
weeks post myocardial infarction event, in control group and experimental
groups.
There was no pertinent differences in
baseline values for the left ventricular ejection fraction, end-diastolic
dimension (in mm), and end-diastolic pressure (Hirata et. al, 2006). The
ejection fraction decreased significantly for all groups except the EPO (0 hr)
group, following the myocardial infarction event (Hirata et. al, 2006). It was
also shown that pressure for the EPO (0 hr) group increased slightly more than
baseline for the end-diastolic pressure, but was still lower than the control
group and other experimental groups (Hirata et. al, 2006).
The same result was observed regarding the
end-diastolic dimension parameter. Relative to the control, the EPO (0 hr)
group is the only group that clearly demonstrates a lower infarct size
percentage at 4 weeks post myocardial infarct event. The EPO (6 hr) and EPO (1
wk) groups’ showed similar infarct sizes to the control group (Hirata et. al,
2006). EPO treatment immediately following the heart attack event, induced by
LAD ligation, was shown to reduce infarct size. Nevertheless, these
size-limiting effects occurs rapidly and is possibly mediated by EPO’s
non-erythropoietic actions such as anti-apoptosis and “scavenging” oxygen
radicals which prevents tissue death (Fisher, 2010).

Figure 7: (A)
Change in left ventricular ejection fraction (LVEDF), (B) end diastolic
dimension, (C) end diastolic pressure, and (D) infarct size during the course
of test period in control group and experimental groups.
In all tested groups, there were no
significant differences observed in hemodynamic parameters such as arterial
blood pressure and heart rate. This finding suggests that EPO actions
selectively affected certain cardiac parameters and did not completely alter
the manner in which the heart functions.
Through this study, it was shown that
erythropoietin can have a cardioprotective effect in dogs and reduce the damage
that results from a heart attack, only if the hormone is administered within a
short time window. It is further suggested that recombinant human EPO may be
used as a supplement for enhancing recovery in patients who have suffered a
myocardial infarction (Hirata et. al, 2006).
Although this study yielded intriguing
results and the investigators clearly tested multiple parameters to support
their conclusions, I believe there are some concerns regarding their
experimental protocols.
Firstly, the entire study was conducted using
47 dogs. For the two experimental protocols; each group, whether a control or
EPO group, only consisted of 6-8 dogs. I believe this sample size is too
restricted for testing an event (heart attack) that can have such very diverse
effects. Furthermore, the investigators state that four dogs were excluded from
the data analysis due to excessive regional myocardial blood flow following the
coronary artery ligation (Hirata et. al, 2006). This further indicates that
certain blood parameters can be very different between dogs, even dogs of the
same breed and relative size.
Secondly, all dogs tested were from the
beagle breed. Evidently, this was done in order to account for any differences
between dog breeds and also because beagles are notably the dog breed most
often used in animal testing, due to their passivity and size (Carlson, 2012). Although
commonly used as surrogates for human testing, I believe the researchers should
have included dogs from different breeds as differences between dog breeds may
also reflect the differences between humans. Also, supplementary testing on
miniature pigs may be appropriate due to the higher organ homology with humans
(Carlson, 2012).
Based on these results, the investigators
suggest that exogenous EPO may be used in long term treatment for its
cardioprotective effects but they do not account for any of the erythropoietic
effects. Using recombinant human erythropoietin to treat ischemia induced
damage may disrupt the production of natural EPO. Lastly, the testing period
was 4 weeks long which may be too short to discount long term effects of EPO
administration.
Despite this, I believe the investigators
carried out their experiments thoroughly and the included results is sufficient
to support the authors’ claims that immediate administration of erythropoietin,
following an ischemia-induced heart attack, may reduce infarct size and promote
neovascularization in the ischemic region.
Future Experiments
In this study, the investigators wanted to
determine whether erythropoietin has cardioprotective effects in ischemic
regions. The results of the study supported their claims, albeit with some
limitations.
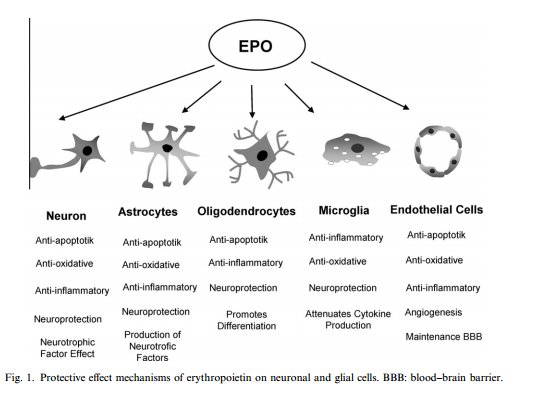
It was previously mentioned that EPO may have
neuroprotective effects; I would like to conduct a study to determine whether
the same cardioprotective effects of EPO can also act as protectants for
neurons in the brain, during ischemia. Ischemic stroke is the condition which
results when the brain does not receive sufficient blood to support its
metabolic requirements (Silverthorn, 2009).

Figure 8:
CT Scan of brain section showing cerebral infarct in the right hemisphere.
For the experiments I would use a rat model,
because of our high understanding of the underlying connections, and I would
perform similar experiments to the ones conducted by Hirata et. al, 2006. By severing
a major cerebral artery in the brain, the resulting ischemia would induce an
infarction (heart attack-like event). Administration of recombinant human EPO
at different time points and concentrations following ligation, will allow us
to monitor the effects at different parameters. EPO can be directly administered
to the ventricles using a cannula implantation, via stereotaxic surgery,
because as a peptide hormone it will be unable to cross the blood brain
barrier.
It has already been implicated that EPO has neovascularization
effects. I expect (and hope) that the increase in capillary formation will be
able to perfuse the ischemic regions and reduce any subsequent necrosis. Using
a rat model will also allow us to use a larger sample size for the study due to
the availability and ethical approval for use in experimentation (Carlson,
2012). A large sample size in a study like this is critical as different rats
(and humans) respond to ischemic events in different ways. These studies will
allow us to better understand the non-erythropoietic effects of erythropoietin
and may also serve as a possible conduit for future treatments.
References
- Carlson, N. R. (2012) Physiology of Behavior (11th
ed.), Pearson Education, Inc. Upper Saddle River, NJ
- Fisher,
J. W. (2010) Landmark advances in the development of erythropoietin. Experimental Biology and Medicine. 235 (12): 1398-1411
- Hirata, A., Minamino, T., Asanuma, H., Fujita, M., Wakeno, M., Myoishi, M., Tsukamoto, O., Okada, K., Koyama, H., Komamura, K., Takashima, S., Shinozaki, Y., Mori, H., Shiraga, M., Kitakaze, M., and Hori, M. (2006) Erythropoietin Enhances
Neovascularization of Ischemic Myocardium and Improves Left Ventricular
Dysfunction after Myocardial Infarction in Dogs. Journal of the American College of Cardiology; 48 (1): 176-184
-
Silverthorn, D. U. (2009). Human Physiology, An integrated approach (5th ed.). Benjamin Cummings,
Pearson Education : San Francisco, CA






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}